The Answer Was Here All the Time: Why Anavex2-73 Can Stop Alzheimer’s Disease
Scientists have spent decades trying to conquer/understand Alzheimer’s disease. Pharmaceutical industry has tried everything under the Sun, from monoclonal antibodies, various nicotinamide derivatives, to a dye named methylene blue. The road is littered with failures. Have we tried too hard and searched too far and wide? Maybe the answer was here all along right in front of our eyes.
The hallmarks of neurodegeneration have been well-established. Numerous proteins form aggregates that include β-amyloid peptides, tau, α-synuclein and others. Whether these protein aggregates cause neurodegeneration is hotly debated among researchers, though sizable portion, if not majority believe they are the consequences instead of the causes today. In addition, there are elevated markers for persistent inflammation in cerebrospinal fluid and peripheral blood¹. In recent years, additional markers for axon degeneration and blood-brain-barrier integrity have been discovered as well. Overall, most scientists today agree that the connections between elevated brain inflammatory factors/responses and neurodegeneration are convincing.
(Lay Summary: neuronal inflammation is the cause of neurodegeneration. Then how can we stop inflammation without comprising immune systems?)
There have been exciting mechanistic studies on memory in recent years. Those studies have identified critical pathways which impair memory formation, neuron health, energy supply, and signal transduction. One of the key discoveries is that neurodegeneration always follows persistent activation of the integrated stress response (ISR, also known as unfolded protein response, UPR)²⁻⁴. Various stresses cause inflammation and activate ISR and over-activation of ISR further produces inflammatory factors in a vicious cycle. The activation of ISR impairs new protein synthesis by phosphorylation of eIF2α⁵. The relationship between new protein synthesis and memory formation, especially long-term memory, is well established⁶⁻⁷. Recent development demonstrates that restoration of protein synthesis in aging mice by a small molecule, ISRIB, restores and enhances memory⁸. Currently ISRIB is in phase II clinical testing. However, there might be potential challenges as ISRIB does not resolve the issue of stresses that activate ISR. One could envision a non-durable response from ISRIB treatment. There are still many potential applications for ISRIB.
(Lay Summary: Neuronal cells in AD patients have diminished ability to synthesize new proteins when needed for memory formation. The likely reason is chronic exposure to inflammation due to aging, neurological diseases, or environment. New molecules such as ISRIB can enable the neurons to synthesize new proteins again despite inflammation when needed for memory. ISRIB works wonders in animal models (Wonderful seminar by Dr. Peter Walter: https://www.youtube.com/watch?v=IsVwHzlp9Lo)).
One pathway seems well agreed upon that causes neuronal inflammation is the TNF-α-induced necroptosis pathway⁹⁻¹³. The pathological causes might vary, which could be chronic infection, genetic predisposition, environmental stresses, or just aging¹⁴. Necroptosis is a cell death pathway with release of proinflammatory factors, as one of the reviews stated “Dying cells fan the flames of inflammation”¹¹. These proinflammatory factors will result in a variety of pathologies that include neurodegeneration. There are numerous players in the necroptosis pathway, from extracellular TNF-α to intracellular kinases that include RIPK1 and RIPK3. There are intense efforts to develop inhibitors targeting RIPK1 and RIPK3 for the treatments of autoimmune diseases and neurodegeneration¹⁵⁻¹⁶ (Denali Therapeutics and Sanofi; Rigel and Eli Lilly, GSK, and many others). Most of these players have multiple functions that include induction of necroptosis. Complete inhibition of RIPK1 or RIPK3 may impact other pathways aside from necroptosis. A recent publication added another critical player in the necroptosis pathway, FKBP12¹⁷. This publication demonstrates that FKBP12, a proline isomerase predominantly expressed in the brain, is essential for the initiation of necroptosis. FKBP12 is extensively studied and is the molecular target for immune suppressants, FK506 and rapamycin. Upon binding of FK506 or rapamycin, the FKBP12-drug complex will inhibit the activities of either calcineurin or mTOR. The immune suppressive functions of these two compounds are due to the inhibition of calcineurin and mTOR respectively¹⁸. FKBP12 itself is a proline isomerase and there are limited efforts in developing pure enzymatic inhibitors of FKBP12. However, recent studies show that inhibition of the enzymatic activities of FKBP12 could be sufficient for the anti-inflammatory functions of FK506 or rapamycin, which makes sense in view of its critical role in necroptosis¹⁷.
(Lay Summary: the major cause of inflammation in neurons is necroptosis, an explosive way of cell death that causes collateral damages. There are many players involved and many companies are developing drugs to reduce necroptosis. So far not much success has been made as majority focuses on RIPK1 and RIPK3. None have targeted FKBP12, as the general perception is that targeting FKBP12 might compromise immune health, which is not true.)
If we inhibit necroptosis, can we stop neurodegeneration? The answer is in plain sight. Monoclonal antibodies against TNF-α are the most prescribed medication in the world. These include Adalimumab (Humira), Etanercept (Enbrel), and Infliximab (Remicade). Millions of patients are taking these medications and the results are convincing: TNF-α sequestration medications reduce the risk of developing Alzheimer’s disease significantly and could be as much as 80% for younger patients¹⁹. Then how about targeting FKBP12 directly? Indeed, patients taking the organ-rejection drug, FK506, rarely develop Alzheimer’s disease at all, if ever²⁰.
(Lay Summary: TNF-α antibodies work wonders for autoimmune diseases and those patients have reduced risk of developing AD by as much as 80%. But antibody drugs require injection, are expensive, and compromise immune system to a certain degree. Still, there are planned clinical trials for FK506 as AD treatment. However, there is always the severe side effect of immune suppression when using FK506 or rapamycin. On the other hand, targeting soluble TNF-α by Inmune Bio should work better than the current antibodies. The impact of anti-TNF drugs on Alzheimer’s were in the news a few years back. https://www.washingtonpost.com/business/economy/pfizer-had-clues-its-blockbuster-drug-could-prevent-alzheimers-why-didnt-it-tell-the-world/2019/06/04/9092e08a-7a61-11e9-8bb7-0fc796cf2ec0_story.html)
These data indicate that targeting necroptosis can stop or at least slowdown the progression of Alzheimer’s disease significantly and inhibition of FKBP12 with a small, non-immune suppressive molecule should be the method of choice. Scientific literature on the functions of FKBP12 lends further support. It has been reported that inhibition of FKBP12 prevents aggregation of β-amyloid peptide, tau, and α-synuclein²¹-²⁴. Furthermore, inhibition of FKBP12 prevented synuclein toxicity²⁵ and restored dopamine concentration, implying future applications in Parkinson disease.
(Lay Summary: a pure FKBP12 inhibitor could block necroptosis and thus neuronal inflammation without compromising immune system. It will stop or slow down neurodegeneration. No one tried because it is assumed that inhibiting FKBP12 would compromise immune system, since FK506 and rapamycin bind to FKBP12 in addition to calcineurin or mTOR. Both drugs are used to treat organ rejection in transplant patients. Inhibition of calcineurin or mTOR leads to immune suppression. Inhibition of FKBP12 alone does not lead to immune suppression but reduction of inflammation from necroptosis. In fact, inhibiting FKBP12 can bring many other benefits for neurons as well! One day most of us old folks will take FKBP12 inhibitors to reduce inflammation and prevent memory loss. We will be as sharp as a 25-year-old when we are 100.)
Inhibition of FKBP12 could alter the course of neurodegenerative disease²⁶. Now there is another question: how much can we reverse the disease, or at least functionally improve memory and cognition? Granted, we will not be able to repair dead neurons or lost neuronal connections. Though, molecules such as ISRIB have shown to improve memory in animal models by restoring new protein synthesis. However, there might be a better target than eIF2α that can relieve ER stress and restore homeostatic states that include protein synthesis. Indeed, Sigma-1 receptor (S1R), residing at mitochondria associated ER membrane (MAM), is a critical ER chaperone that prevents over activation of ISR²⁷⁻³³. S1R is regarded as a good target for treating neurological disorders³⁴. It is also interesting to note that S1R might be responsible for the folding and processing of α-synuclein and β-amyloid and FKBP12 is responsible for their aggregation. Consequently sigma-1 receptor agonists have been shown to be effective in treating neurological disorders in animal models³⁵ that include ALS²⁹, fragile X syndrome³⁶, Rett syndrome³⁷, and VWM leukodystrophy³⁸.
The combination of activation of sigma-receptor and inhibition of FKBP12 could change the course of Alzheimer’s disease. S1R agonism can restore memory to certain extent and FKBP12 antagonism can slow down disease progression by eliminating the cause of degeneration, necroptosis induced inflammation. If a single molecule can do both, that drug could have a far-reaching impact on AD treatment.
(Lay Summary: how about one found out he/she already has significant memory impairment? Stop the progression with a FKBP12 inhibitor is not good enough! Then we need S1R agonist that can repair mitochondria dysfunction and restore protein synthesis. Thus, memory and cognition can improve significantly. A S1R agonist and a FKBP12 inhibitor combination might do wonders for AD patients as well as an aging population in general!)
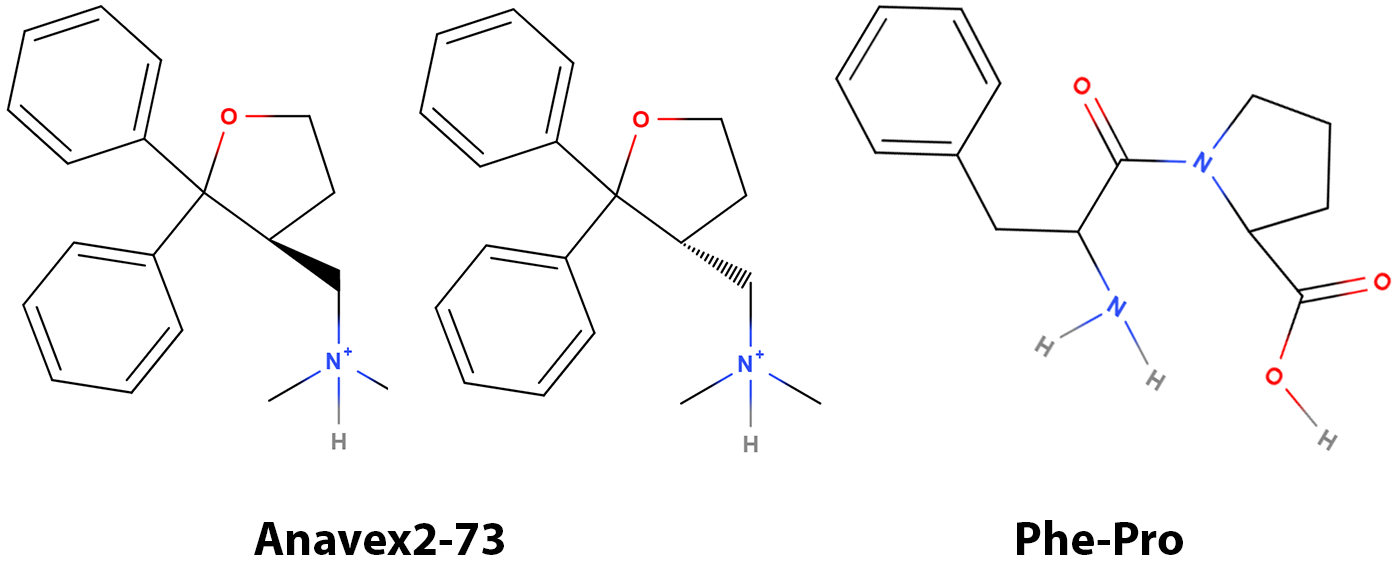
Figure 1. Molecular structures of Anavex2-73 and Phe-Pro dipeptide. Anavex2-73 is presumably a racemic mixture of two enantiomers.
A small molecule in clinical development has come into view in the last two years, named Anavex2-73 (Figure 1). It has shown impressive safety records in early stages of clinical trials. Anavex2-73 is a S1R agonist with moderate affinity and has produced impressive clinical results from double-blinded studies treating Rett syndrome and Parkinson disease dementia. Currently it is in a phase II/III trial for Alzheimer’s disease. However, neither the scientific nor the investment communities are fully endorsing the clinical results or the potential in AD treatment. Granted, there are thousands of agonists of S1R with higher affinities than that of Anavex2-73 and none of them have been proven to have significant clinical applications. Is Anavex2-73 unique?

Figure 2. Possible binding mode of Anavex2-73 to the active site of FKBP12.
Agonism to S1R will provide temporary relief of ER stress (ISR) and we can expect short-term improvement in symptoms resulting from AD. However, a sustained response requires blunting the source of inflammation, necroptosis. From a structural perspective, Anavex2-73 features a phenyl group (Benzene) adjacent to a 5-membered ring, a structural similarity to that of Phe-Pro dipeptide. FKBP12 recognizes and prefers a substrate with sequence containing Phe-Pro for catalyzing the isomerization of the corresponding peptide bond³⁹. We suspect that Anavex2-73 is an inhibitor of FKBP12 and subsequently conducted preliminary docking experiments. The results indicate that Anavex2-73 might be an inhibitor of FKBP12 (Figure 2) with reasonable affinity, likely in the nanomolar range. Granted, wet lab experiments are needed to confirm or disprove the docking studies, as docking alone does produce artifacts. However, the fit to the active site is intriguing, especially the impressive hydrophobic interactions between Anavex2-73 and the active site without steric strains. Further laboratory investigation is warranted to obtain binding affinities. Maybe it is too condescending to ask “Did Anavex stumble onto the right compound”?

Figure 3. Comparison of possible binding modes of Anavex2-73 to that of rapamycin
Anavex2-73 could bind to the active site of FKBP12 without engagement with either mTOR or calcineurin (Figure 3). Therefore, it will not be immune suppressive in long-term use.
(Lay Summary: There are thousands of S1R agonists around. Why Anavex2-73 seems to work better. Our modeling studies suggest that Anavex2-73 is unique in that it could also be a potent FKBP12 inhibitor, even Anavex (the company) is not aware of it! Most likely Anavex2-73 will succeed in clinical trial in AD because it works in both pathways!)
Have we finally found a compound that can do both, activate S1R and inhibit FKBP12? Is there light at the end of the tunnel in AD drug development? All of us are waiting with abated breath for the trial results from Anavex. Maybe, just maybe, we can relax knowing the mechanisms of Anavex2-73. The possible dual actions of Anavex2-73 might project altered course of disease progression as well as improved memory and cognition for AD patients.
(Lay Summary: Upon laboratory confirmation of the modeling studies, Anavex2-73 could change the way we treat or see Alzheimer’s disease. It could restore memory function and completely change the trajectory of disease progression. As a matter of fact, it should work on other diseases as well such as Parkinson’s, ALS, Fragile X syndrome, Rett, and possibly others. As for the ongoing trials, the results could be so spectacular that we would have never expected!)
Disclosure: I/we have a beneficial long position in the shares of AVXL. I have no business relationship with any company mentioned in this article. I am not a financial advisor, and this article is not financial advice.
References
- Cullen NC, Malarstig AN, Stomrud E, Hansson O, Mattsson-Carlgren N. Accelerated inflammatory aging in Alzheimer's disease and its relation to amyloid, tau, and cognition. Sci Rep. 2021;11(1):1965. Epub 20210121. doi: 10.1038/s41598-021-81705-7. PubMed PMID: 33479445; PMCID: PMC7820414.
- Bond S, Lopez-Lloreda C, Gannon PJ, Akay-Espinoza C, Jordan-Sciutto KL. The Integrated Stress Response and Phosphorylated Eukaryotic Initiation Factor 2alpha in Neurodegeneration. J Neuropathol Exp Neurol. 2020;79(2):123-43. doi: 10.1093/jnen/nlz129. PubMed PMID: 31913484; PMCID: PMC6970450.
- Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368(6489). doi: 10.1126/science.aat5314. PubMed PMID: 32327570; PMCID: PMC8997189.
- Korneeva NL. Integrated Stress Response in Neuronal Pathology and in Health. Biochemistry (Mosc). 2022;87(Suppl 1):S111-S27. doi: 10.1134/S0006297922140103. PubMed PMID: 35501991.
- Sharma V, Sood R, Khlaifia A, Eslamizade MJ, Hung TY, Lou D, Asgarihafshejani A, Lalzar M, Kiniry SJ, Stokes MP, Cohen N, Nelson AJ, Abell K, Possemato AP, Gal-Ben-Ari S, Truong VT, Wang P, Yiannakas A, Saffarzadeh F, Cuello AC, Nader K, Kaufman RJ, Costa-Mattioli M, Baranov PV, Quintana A, Sanz E, Khoutorsky A, Lacaille JC, Rosenblum K, Sonenberg N. eIF2alpha controls memory consolidation via excitatory and somatostatin neurons. Nature. 2020;586(7829):412-6. Epub 20201007. doi: 10.1038/s41586-020-2805-8. PubMed PMID: 33029011; PMCID: PMC7874887.
- Jarome TJ, Helmstetter FJ. Protein degradation and protein synthesis in long-term memory formation. Front Mol Neurosci. 2014;7:61. Epub 20140626. doi: 10.3389/fnmol.2014.00061. PubMed PMID: 25018696; PMCID: PMC4072070.
- Alkon DL, Epstein H, Kuzirian A, Bennett MC, Nelson TJ. Protein synthesis required for long-term memory is induced by PKC activation on days before associative learning. Proc Natl Acad Sci U S A. 2005;102(45):16432-7. Epub 20051028. doi: 10.1073/pnas.0508001102. PubMed PMID: 16258064; PMCID: PMC1283453.
- Krukowski K, Nolan A, Frias ES, Boone M, Ureta G, Grue K, Paladini MS, Elizarraras E, Delgado L, Bernales S, Walter P, Rosi S. Small molecule cognitive enhancer reverses age-related memory decline in mice. Elife. 2020;9. Epub 20201201. doi: 10.7554/eLife.62048. PubMed PMID: 33258451; PMCID: PMC7721440.
- Zhang S, Tang MB, Luo HY, Shi CH, Xu YM. Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis. 2017;8(6):e2905. Epub 20170629. doi: 10.1038/cddis.2017.286. PubMed PMID: 28661482; PMCID: PMC5520937.
- Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. 2019;4(15). Epub 20190808. doi: 10.1172/jci.insight.128834. PubMed PMID: 31391333; PMCID: PMC6693822.
- Newton K, Dixit VM, Kayagaki N. Dying cells fan the flames of inflammation. Science. 2021;374(6571):1076-80. Epub 20211125. doi: 10.1126/science.abi5934. PubMed PMID: 34822265.
- Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20(1):19-33. doi: 10.1038/s41583-018-0093-1. PubMed PMID: 30467385; PMCID: PMC6342007.
- Jayaraman A, Htike TT, James R, Picon C, Reynolds R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer's disease hippocampus. Acta Neuropathol Commun. 2021;9(1):159. Epub 20210928. doi: 10.1186/s40478-021-01264-w. PubMed PMID: 34625123; PMCID: PMC8501605.
- Thadathil N, Nicklas EH, Mohammed S, Lewis TL, Jr., Richardson A, Deepa SS. Necroptosis increases with age in the brain and contributes to age-related neuroinflammation. Geroscience. 2021;43(5):2345-61. Epub 20210913. doi: 10.1007/s11357-021-00448-5. PubMed PMID: 34515928; PMCID: PMC8599532.
- Martens S, Hofmans S, Declercq W, Augustyns K, Vandenabeele P. Inhibitors Targeting RIPK1/RIPK3: Old and New Drugs.Trends Pharmacol Sci. 2020;41(3):209-24. Epub 20200205. doi: 10.1016/j.tips.2020.01.002. PubMed PMID: 32035657.
- Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, Belfiore R, Winslow W, Oddo S. Necroptosis activation in Alzheimer's disease. Nat Neurosci. 2017;20(9):1236-46. Epub 20170724. doi: 10.1038/nn.4608. PubMed PMID: 28758999.
- Wang Z, Feng J, Yu J, Chen G. FKBP12 mediates necroptosis by initiating RIPK1-RIPK3-MLKL signal transduction in response to TNF receptor 1 ligation. J Cell Sci. 2019;132(10). Epub 20190520. doi: 10.1242/jcs.227777. PubMed PMID: 31028177.
- Tong M, Jiang Y. FK506-Binding Proteins and Their Diverse Functions. Curr Mol Pharmacol. 2015;9(1):48-65. doi: 10.2174/1874467208666150519113541. PubMed PMID: 25986568; PMCID: PMC6611466.
- Zhou M, Xu R, Kaelber DC, Gurney ME. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer's disease in patients with rheumatoid arthritis and psoriasis. PLoS One. 2020;15(3):e0229819. Epub 20200323. doi: 10.1371/journal.pone.0229819. PubMed PMID: 32203525; PMCID: PMC7089534.
- Taglialatela G, Rastellini C, Cicalese L. Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors. J Alzheimers Dis. 2015;47(2):329-33. doi: 10.3233/JAD-150065. PubMed PMID: 26401556; PMCID: PMC4923720.
- Caminati G, Martina MR, Menichetti S, Procacci P. Blocking the FKBP12 induced dendrimeric burst in aberrant aggregation of alpha-synuclein by using the ElteN378 synthetic inhibitor. J Enzyme Inhib Med Chem. 2019;34(1):1711-5. doi: 10.1080/14756366.2019.1667342. PubMed PMID: 31547734; PMCID: PMC6764402.
- Deleersnijder A, Van Rompuy AS, Desender L, Pottel H, Buee L, Debyser Z, Baekelandt V, Gerard M. Comparative analysis of different peptidyl-prolyl isomerases reveals FK506-binding protein 12 as the most potent enhancer of alpha-synuclein aggregation. J Biol Chem. 2011;286(30):26687-701. Epub 20110607. doi: 10.1074/jbc.M110.182303. PubMed PMID: 21652707; PMCID: PMC3143632.
- Gerard M, Deleersnijder A, Daniels V, Schreurs S, Munck S, Reumers V, Pottel H, Engelborghs Y, Van den Haute C, Taymans JM, Debyser Z, Baekelandt V. Inhibition of FK506 binding proteins reduces alpha-synuclein aggregation and Parkinson's disease-like pathology. J Neurosci. 2010;30(7):2454-63. doi: 10.1523/JNEUROSCI.5983-09.2010. PubMed PMID: 20164329; PMCID: PMC6634531.
- Hong HS, Hwang JY, Son SM, Kim YH, Moon M, Inhee MJ. FK506 reduces amyloid plaque burden and induces MMP-9 in AbetaPP/PS1 double transgenic mice. J Alzheimers Dis. 2010;22(1):97-105. doi: 10.3233/JAD-2010-100261. PubMed PMID: 20847451.
- Caraveo G, Soste M, Cappelleti V, Fanning S, van Rossum DB, Whitesell L, Huang Y, Chung CY, Baru V, Zaichick S, Picotti P, Lindquist S. FKBP12 contributes to alpha-synuclein toxicity by regulating the calcineurin-dependent phosphoproteome. Proc Natl Acad Sci U S A. 2017;114(52):E11313-E22. Epub 20171211. doi: 10.1073/pnas.1711926115. PubMed PMID: 29229832; PMCID: PMC5748183.
- Caminati G, Procacci P. Mounting evidence of FKBP12 implication in neurodegeneration. Neural Regen Res. 2020;15(12):2195-202. doi: 10.4103/1673-5374.284980. PubMed PMID: 32594030; PMCID: PMC7749462.
- Rosen DA, Seki SM, Fernandez-Castaneda A, Beiter RM, Eccles JD, Woodfolk JA, Gaultier A. Modulation of the sigma-1 receptor-IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci Transl Med. 2019;11(478). doi: 10.1126/scitranslmed.aau5266. PubMed PMID: 30728287; PMCID: PMC6936250.
- Ha Y, Dun Y, Thangaraju M, Duplantier J, Dong Z, Liu K, Ganapathy V, Smith SB. Sigma receptor 1 modulates endoplasmic reticulum stress in retinal neurons. Invest Ophthalmol Vis Sci. 2011;52(1):527-40. Epub 20110125. doi: 10.1167/iovs.10-5731. PubMed PMID: 20811050; PMCID: PMC3053296.
- Lee PT, Lievens JC, Wang SM, Chuang JY, Khalil B, Wu HE, Chang WC, Maurice T, Su TP. Sigma-1 receptor chaperones rescue nucleocytoplasmic transport deficit seen in cellular and Drosophila ALS/FTD models. Nat Commun. 2020;11(1):5580. Epub 20201104. doi: 10.1038/s41467-020-19396-3. PubMed PMID: 33149115; PMCID: PMC7642387.
- Hong J, Wang L, Zhang T, Zhang B, Chen L. Sigma-1 receptor knockout increases alpha-synuclein aggregation and phosphorylation with loss of dopaminergic neurons in substantia nigra. Neurobiol Aging. 2017;59:171-83. Epub 20170810. doi: 10.1016/j.neurobiolaging.2017.08.007. PubMed PMID: 28870519.
- Bernard-Marissal N, Medard JJ, Azzedine H, Chrast R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain. 2015;138(Pt 4):875-90. Epub 20150211. doi: 10.1093/brain/awv008. PubMed PMID: 25678561.
- Ha Y, Saul A, Tawfik A, Zorrilla EP, Ganapathy V, Smith SB. Diabetes accelerates retinal ganglion cell dysfunction in mice lacking sigma receptor 1. Mol Vis. 2012;18:2860-70. Epub 20121130. PubMed PMID: 23233788; PMCID: PMC3519370.
- Omi T, Tanimukai H, Kanayama D, Sakagami Y, Tagami S, Okochi M, Morihara T, Sato M, Yanagida K, Kitasyoji A, Hara H, Imaizumi K, Maurice T, Chevallier N, Marchal S, Takeda M, Kudo T. Fluvoxamine alleviates ER stress via induction of Sigma-1 receptor. Cell Death Dis. 2014;5(7):e1332. Epub 20140717. doi: 10.1038/cddis.2014.301. PubMed PMID: 25032855; PMCID: PMC4123092.
- Salaciak K, Pytka K. Revisiting the sigma-1 receptor as a biological target to treat affective and cognitive disorders. Neurosci Biobehav Rev. 2022;132:1114-36. Epub 20211101. doi: 10.1016/j.neubiorev.2021.10.037. PubMed PMID: 34736882; PMCID: PMC8559442.
- Bogar F, Fulop L, Penke B. Novel Therapeutic Target for Prevention of Neurodegenerative Diseases: Modulation of Neuroinflammation with Sig-1R Ligands. Biomolecules. 2022;12(3). Epub 20220225. doi: 10.3390/biom12030363. PubMed PMID: 35327555; PMCID: PMC8945408.
- Reyes ST, Deacon RMJ, Guo SG, Altimiras FJ, Castillo JB, van der Wildt B, Morales AP, Park JH, Klamer D, Rosenberg J, Oberman LM, Rebowe N, Sprouse J, Missling CU, McCurdy CR, Cogram P, Kaufmann WE, Chin FT. Effects of the sigma-1 receptor agonist blarcamesine in a murine model of fragile X syndrome: neurobehavioral phenotypes and receptor occupancy. Sci Rep. 2021;11(1):17150. Epub 20210825. doi: 10.1038/s41598-021-94079-7. PubMed PMID: 34433831; PMCID: PMC8387417.
- Kaufmann WE, Sprouse J, Rebowe N, Hanania T, Klamer D, Missling CU. ANAVEX(R)2-73 (blarcamesine), a Sigma-1 receptor agonist, ameliorates neurologic impairments in a mouse model of Rett syndrome. Pharmacol Biochem Behav. 2019;187:172796. Epub 20191105. doi: 10.1016/j.pbb.2019.172796. PubMed PMID: 31704481.
- Atzmon A, Herrero M, Sharet-Eshed R, Gilad Y, Senderowitz H, Elroy-Stein O. Drug Screening Identifies Sigma-1-Receptor as a Target for the Therapy of VWM Leukodystrophy. Front Mol Neurosci. 2018;11:336. Epub 20180918. doi: 10.3389/fnmol.2018.00336. PubMed PMID: 30279648; PMCID: PMC6153319.
- Jakob RP, Zoldak G, Aumuller T, Schmid FX. Chaperone domains convert prolyl isomerases into generic catalysts of protein folding. Proc Natl Acad Sci U S A. 2009;106(48):20282-7. Epub 20091117. doi: 10.1073/pnas.0909544106. PubMed PMID: 19920179; PMCID: PMC2787138.